本文华算科技将从几何定义、电子与键合机制、稳定性与吸附能以及在具体催化反应中的意义等多个维度,对这两个核心概念进行系统性比较和深入剖析。
几何结构与对称性定义
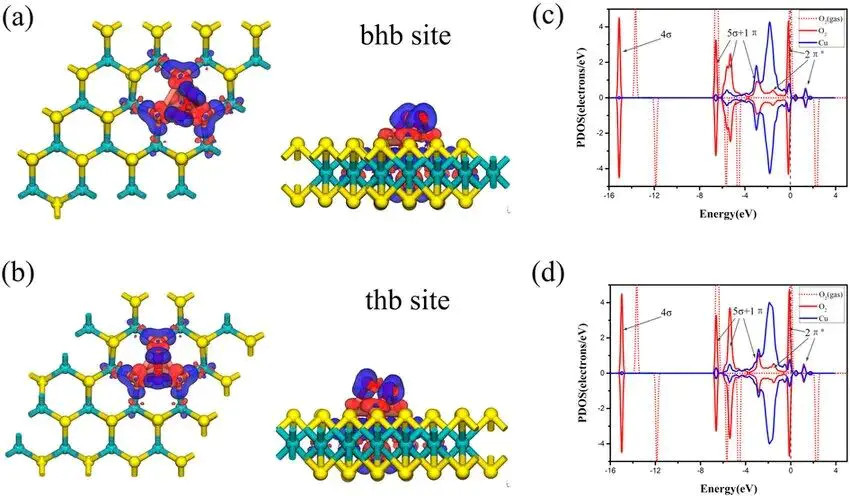
顶位桥位顶位(:。在这种构型下,吸附原子与表面形成了“一对一”的垂直键合关系。例如,在常见的面心立方(fcc)金属的(111)晶面上,顶位具有较高的局部对称性,其点群通常为C3v桥位(:。吸附原子与这两个金属原子大致处于同一个平面内,形成了一个“一对二”的键合结构。在fcc(111)晶面上,桥位的局部对称性低于顶位,其点群通常为C2v除了这两种基本构型,金属表面还存在Hollow Site)值得注意的是,尽管这些概念在文献中被广泛使用,但学术界并未给出一个统一的、基于精确原子坐标的“标准数学定义”。研究中通常通过第一性原理计算在不同位点的精确坐标和键长、键角等参数。
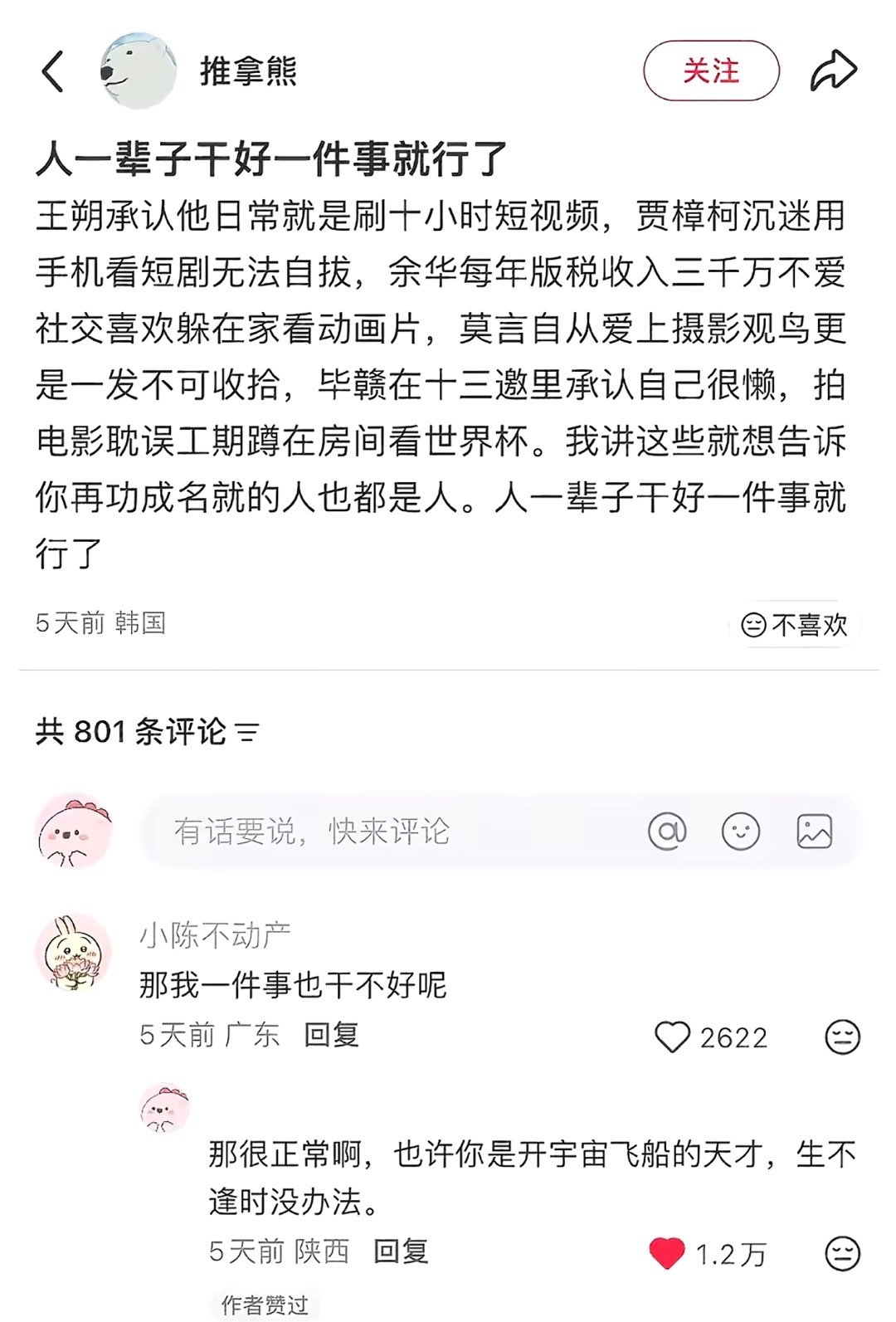
电子结构与键合机制
相互作用的核心顶位键合机制在顶位吸附时,吸附质的成键轨道主要与正下方的单个金属原子的d轨道和s轨道发生最强烈的相互作用,形成一个局域性很强的σ型化学键。
。
:。吸附质的轨道会同时与两个相邻金属原子的d轨道发生交叠。这种更广泛的轨道杂化使得成键电子能够在一个更大的范围内离域,从而可能形成更稳定的化学键。
是桥位键合稳定性的重要来源。
吸附能,其绝对值越大(即数值越负),代表吸附越稳定。
,即具有更高的吸附能。这是因为在桥位,吸附原子可以与两个表面原子成键,形成了更高的配位数,从而获得了更大的能量释放。例如,研究表明1,5-三唑分子在Cu(111)表面的吸附中,桥位模式能量最低(最稳定),而顶位模式能量最高(最不稳定)。同样,O₂在α-MnO₂表面的桥位吸附能(-2.8777 eV)也显著高于顶位(-1.2242 eV)。
吸附质与基底的化学本质不同的吸附质-基底组合表现出不同的偏好。例如,O原子在Ru(0001)表面,三重hcp位点比双重桥位更稳定,而顶位则完全不稳定。
:表面应力与重构表面的应变状态也会影响不同位点的相对稳定性。
10.1039/D4EN00953C
结论
与作为金属表面最基本的两种吸附构型,其差异贯穿于表面科学和多相催化的各个层面。
上看,它们代表了“一对一”与“一对二”的原子配位模式;从电子与键合机制上看,桥位通常因更高的配位数而提供更强的吸附,但这受到覆盖度和体系化学本质的精细调控。
中被放大,通过影响反应中间体的稳定性和过渡态能垒,直接决定了催化剂的活性与选择性。