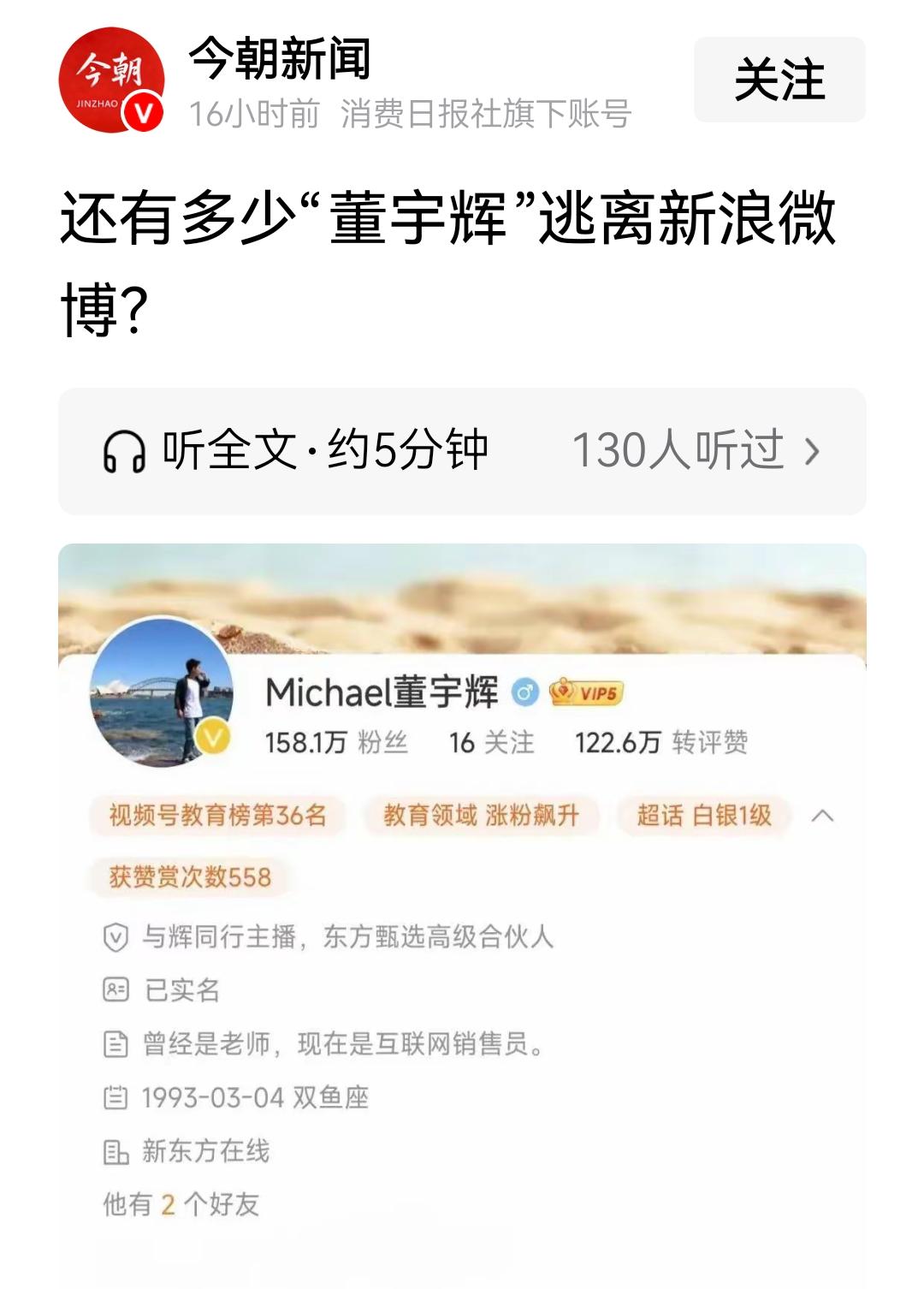
在、、以及有机反应机理研究中,酸性位点的类型与强度是决定反应路径与产物分布的核心因素。Brønsted酸与Lewis酸位点是两种最为基础且普遍存在的酸性类型,其区别不仅体现在质子供受机理上,更影响催化剂与反应物之间的电子相互作用模式。
通常表现为质子供体,能够通过质子化反应活化底物;则是电子对受体,通过与底物电子对配位形成新的反应活性中心。
在理论计算研究中,对两类酸位点的区分和表征是理解催化机理的重要前提,而在实验研究中,常通过吡啶红外光谱(₃本文将系统阐述Brønsted酸与Lewis酸的基本定义、电子结构特征、理论与实验表征方法、在催化反应中的作用以及在论文中常见的分析策略,并结合近年来高水平研究案例进行深入解析。
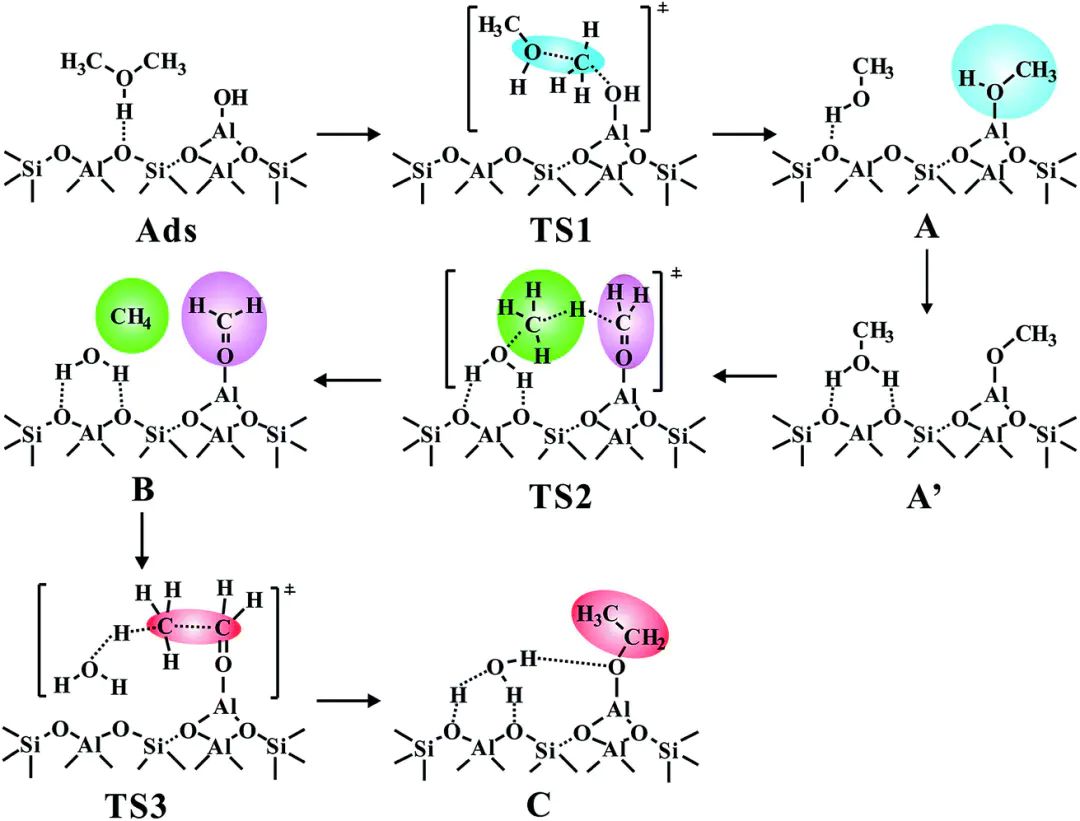
Brønsted酸的定义源自Brønsted–Lowry酸碱理论,即任何能够提供质子(HBrønsted酸当底物分子接近Brønsted酸位点时,可以接受该质子,从而改变自身的电荷分布与几何构型,降低反应能垒。这一机制在烯烃异构化、烃类裂解、烷基化等酸催化反应中尤为重要。不仅取决于其周围骨架的电子环境,也与密切相关。

。在固体表面,这类酸性位点通常是具有未饱和配位的金属离子(如Al³TiZrLewis酸位点与底物分子相互作用的方式主要是通过接受其孤对电子形成配位络合物,这在羰基化合物的活化、烯烃环加成以及某些氧化反应中起到关键作用。
改变电子分布在选择性控制上具有独特优势。
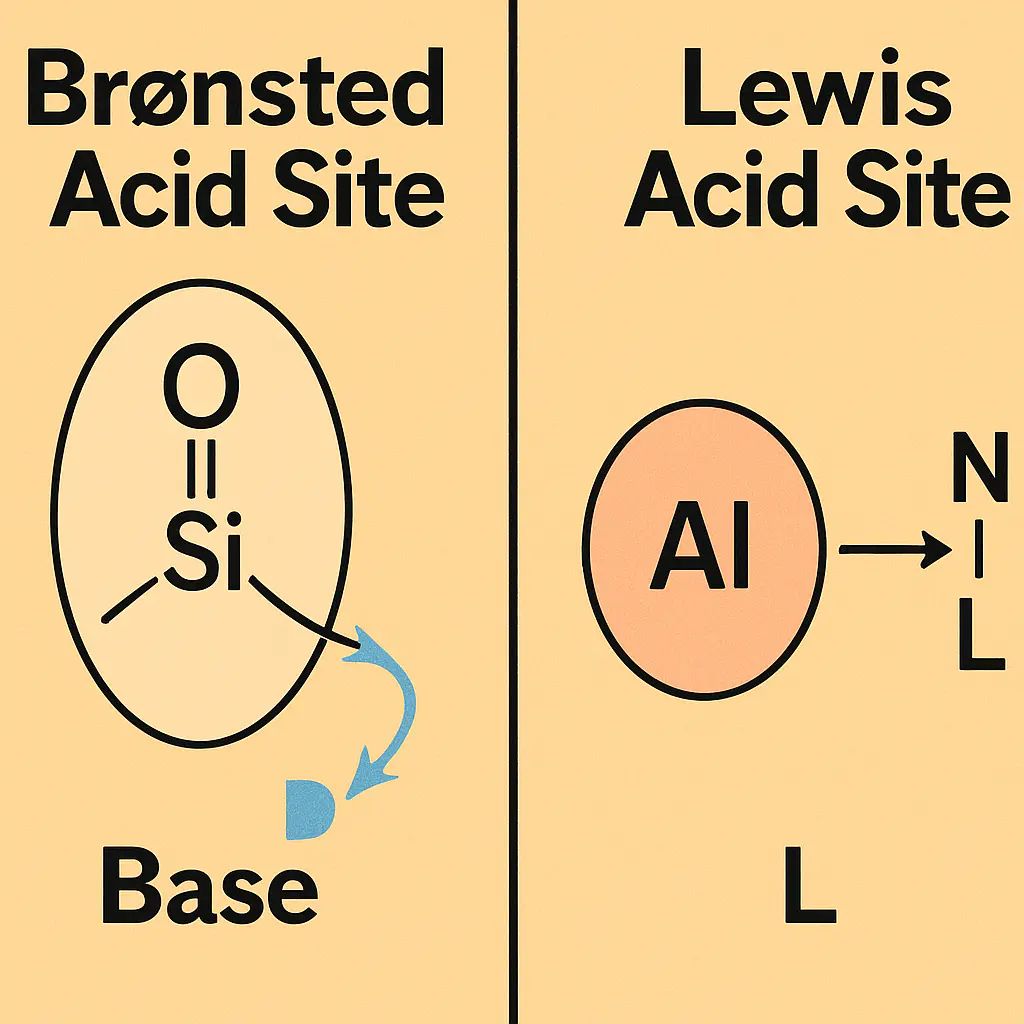
从的角度看,Brønsted酸位点的酸性起源在于羟基中的质子易于离去,其离解能受邻近骨架原子电负性与局域电子密度分布影响。
在中,可通过计算O–H键的断裂能、质子化能或使用差分电荷密度分析来量化Brønsted酸强度。

Lewis酸位点的电子结构特征主要体现在金属中心的空轨道可用于接受底物的孤电子对⁴⁺、⁴⁺等高价金属具有更强的在第一性原理计算中,Lewis酸性可通过分析金属原子的来加以表征。这类分析在催化反应机理研究中极具解释力。
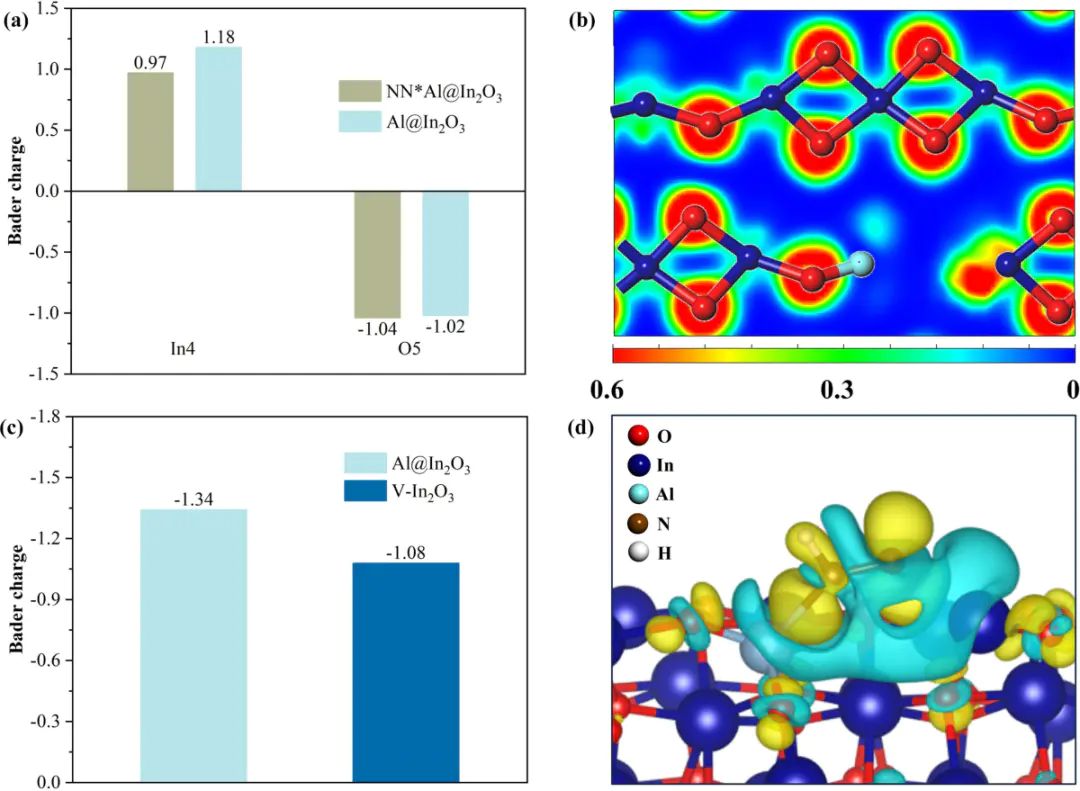
理论计算中酸性位点的表征与分析
常用的分析方法包括。此外,还可以利用振动频率分析比较羟基质子化前后的特征峰位变化,模拟实验中的红外光谱结果。
Lewis酸位点的理论表征则主要依赖于。通过比较配位前后的金属态密度,可以直接观察底物孤电子对与金属空轨道之间的相互作用。
分子轨道(HOMO/LUMO)在高水平论文中,往往会将这两类酸位点的计算结果与实验Py-IR光谱中吡啶分子在1540 cmBrønsted)与1450 cmLewis)处的吸收峰对应起来,以实现理论与实验的DOI:10.1039/D3RE00007A
实验方法与论文中的分析策略
Brønsted酸与Lewis酸的区分常依赖于,因吡啶分子在与Brønsted酸位点结合时会被质子化,产生特征吸收峰,而与Lewis酸位点结合时则以配位络合的方式存在,产生另一类吸收峰。
氨程序升温脱附(NH-TPD)在高水平论文中,研究者常将。例如在这类论文的分析策略通常包括三步:第一步通过理论计算预测酸性变化趋势;第二步利用Py-IR与NH-TPD等实验手段定量分析酸性类型与浓度;第三步将酸性参数与反应动力学数据进行相关性分析,从而催化反应中的功能作用与协同效应
质子化反应物DOI:Lewis酸位点则更多参与电子结构的重构与底物配位。例如在醛酮缩合反应中,Lewis酸位点通过与羰基氧配位,降低C=O键的LUMO能级,从而促进亲核加成过程。
Brønsted与Lewis酸位点可以协同作用结论与展望
。从电子结构到反应机理,这两类酸位点在理论计算与实验表征中都有明确的分析方法。
未来的发展方向可能包括原位光谱与实时计算相结合,实现反应过程中的酸性动态演化追踪;利用加速酸性位点结构–性能关系的预测;以及在多功能催化剂设计中精准调控Brønsted与Lewis酸比例,以适配复杂多步反应的需求。