华算科技DFTMDMC区分 DFT、MD、MC,先看它们描述的物理对象。DFT 从电子密度和原子构型出发,计算得到体系在给定结构下的能量、力和电子结构;MD 从原子位置和速度出发,沿时间演化得到原子轨迹;MC 从概率权重出发,在可能构型或事件之间抽样。三者均可用于材料计算研究,但它们并不存在“精度从低到高”的线性关系。
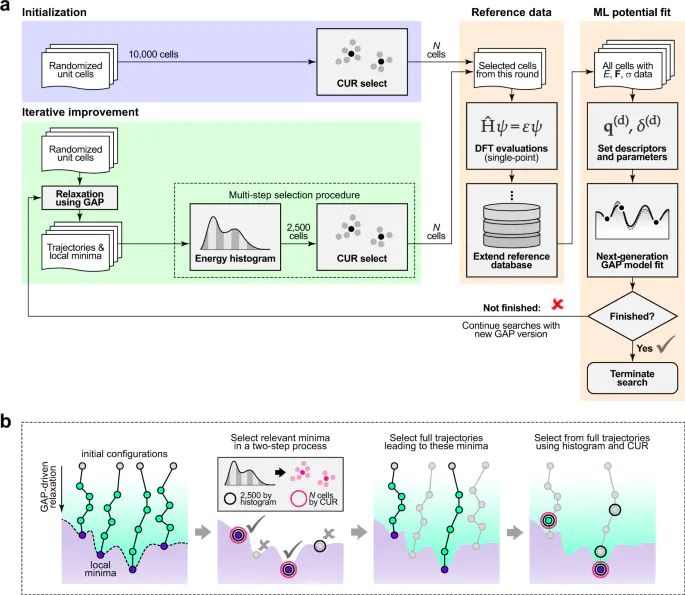
图1. JARVIS 按长度和时间尺度整理计算材料设计方法,显示 DFT、MD、介观模型和连续模型各自适合的尺度范围。DOI:10.1038/s41524-020-00440-1
同一个缩写在不同语境中可能有不同含义。比如 MD 可以指经典,也可以指 AIMD;MC 可以指平衡态 Metropolis 抽样,也可以指 GCMC 或 kMC;DFT 可以作为单点能量计算,也可以给轨迹、势函数或统计模型提供输入。解读方法名称时需要同时明确其研究对象、系综、时间尺度和输出量,这样才不会把不同层级的结果混用。
二、DFT为什么更像电子结构能量尺?
密度泛函DFT 结果通常需要明确参考态。比如形成能要说明元素参考相,吸附能要说明清洁表面和孤立分子,缺陷形成能要说明化学势和电荷态。DFT 数值的物理意义来自“最终构型”和“参考构型”的能量差,不是某个总能量本身。新手如果只比较 OUTCAR 里的总能量,很容易混淆体系大小、元素数目和参考态的差异。
DFT 更擅长回答“这个构型的电子结构和能量怎样”,但它不能直接给出长时间尺度的扩散行为、真实合成过程或大规模相分离过程。DFT 可以驱动 AIMD,也可以为机器学习势或 Monte Carlo 模型提供能量标签;这时 DFT 扮演的是高精度能量标尺,而非能够无限扩展的轨迹计算工具。
从势能面角度看,一次 DFT 计算像给某个构型标定一个能量高度,结构优化相当于沿局部势能面找到附近的极小值点。当研究目标落在电子结构、键合强弱、能量差和反应路径上,DFT 通常是第一层方法。但如果目标转向纳秒级扩散、几十万构型的平衡分布或孔道吸附统计,单靠 DFT 计算成本往往过高。
三、MD为什么更像原子运动轨迹?
MD 与 DFT 的差别并非仅仅是计算速度差异。MD 关心有限温度下结构如何随时间演化,所以它适合分析扩散通道、界面水层、熔融、相变萌生、孔道迁移和局部配位重排。时间步长、系综、温控方式、势函数质量和模拟时长,都会改变轨迹能覆盖的构型范围。

MD 轨迹本身还需要转化成统计量,例如 RDF 描述局部配位,MSD 描述平均位移增长,扩散系数来自长时间斜率,能量和温度曲线用于检查体系是否达到目标状态。单张结构快照只能反映某一时刻的构型,不能直接代表体系的整体动力学行为。
MD 也有其局限性。经典 MD 的可靠性取决于势函数能否描述目标化学环境,AIMD 的成本又限制了体系大小和模拟时长。扩散、重构、溶剂化和有限温稳定性这类问题常用 MD,但还要让时间尺度、采样次数和势函数适用范围匹配研究目标。
四、MC为什么更像热力学抽样器?
在材料计算中,MC 常用于合金有序-无序、相分离、吸附覆盖度、GCMC 吸附等温线、缺陷分布和 kMC 事件演化。MC step 不等于 fs 或 ps,普通 Metropolis MC 主要给平衡构型分布;kMC 在已知事件速率时可以连接时间尺度。这一特点决定了 MC 适合回答“哪些状态更常出现”,而不是直接展示原子如何振动。
MC 的优势在于大构型空间和热力学权重。多组分合金中,元素排布数量极大,逐个枚举几乎不可行;MC 可以通过接受准则将采样重点集中在热力学上概率更高的区域。吸附体系中,GCMC 允许粒子数涨落,适合描述孔材料在给定温度和化学势下的吸附量。
MC 也依赖能量模型。如果能量函数、簇展开、相互作用参数或事件速率不可靠,MC 只会高效得到错误模型的计算结果。因此,很多 MC 工作会先用 DFT 计算构型能量,再用 MC 扩展到温度、组成和大尺度构型分布。这不是把 DFT 替换掉,而是将 DFT 难以直接覆盖的采样空间交由统计方法处理。