华算科技DFT 计算带隙计算带隙Kohn−Sham不同实验测到的是光学带隙、准粒子带隙或激子相关能量,物理含义并不完全相同。
隙和间接带隙。最小间接带隙,却拿来与光吸收外推的直接跃迁比较,即使方法本身合理,也会出现看似异常的偏差。
典型半导体中,PBE 对 Si 的带隙约 0.6 eV,而室温实验间接带隙约 1.1 eV;对 ZnO,PBE 可能给出约 0.7–1.0 eV,而实验值约 3.3 eV。这类偏差不是个别算例异常,而是近似泛函的系统问题。
式中 εCBM 是导带底 Kohn−Sham 本征值,εVBM 是价带顶本征值。这个差值常用于材料筛选,但它不是严格意义上的准粒子带隙。
另一个常PBE 往往略微高估晶格常数,而带隙对键长和键角很敏感;压缩或拉伸 1% 的晶格常数,就可能让某些半导体带隙变化几十到数百 meV。中既有电子结构近似,也可能夹杂结构误差。
真实电子体系在电子数跨过整数时,交换关联势会发生跃迁;LDA 和 GGA 往往无法正确描述这一跃迁,因此导带位置被低估。
这里 I 是电离能,A 是电子亲和能,E(N) 是 N 电子体系总能。严格基态 DFT 的基本带隙应由总能差给出,而普通本征值差缺少交换关联导数不连续项。
自旋轨道耦合含 Bi、Pb、I、Te 等重元素的材料中,SOC 可能把带隙降低 0.1–1 eV,钙钛矿和拓扑材料尤其明显。若实验讨论的是含 SOC 的真实能级,而计算只做标量相对论近似,偏差不能完全归因于 PBE。
更合理的策略是把 PBE 带隙作为低成本描述符,而不是最终实验预测值;进入候选名单后,再用 HSE、GW 或实验标定模型提高精度。
HSE 通常能显著改善半导体带隙,但计算成本高于 PBE,且 25% 精确交换并非所有材料都最优。GW 更接近准粒子能级,但依赖起始波函数、空带数和介电矩阵截断。对于大规模筛选,可先用 PBE 判断趋势,再对候选材料做高精度复算。

缺陷体系尤其需要谨慎。若论文讨论深能级缺陷或发光中心,单纯 PBE 能带图通常不足以支撑结论。
实验也不唯一
实验值并不是固定常数。多数半导体带隙随温度升高而减小,零温计算结果与室温吸收谱之间本来就存在电子−声子耦合和热膨胀差异。
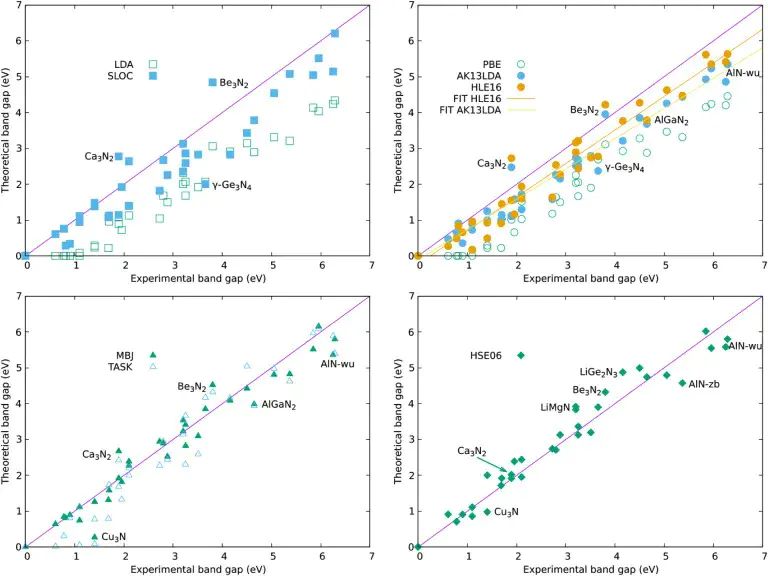
图4:氮化物带隙基准测试展示不同 DFT 近似对半导体带隙预测的偏差。DOI:10.1038/s41524-026-02009-w。
若需要给出与实验更一致的数值,可以采用分级验证:先用 PBE 优化结构,再用 HSE 或 GW 做单点能级;对强激子材料进一步结合 BSE 计算光学吸收。这样既控制成本,也能区分结构误差、准粒子修正和激子效应。
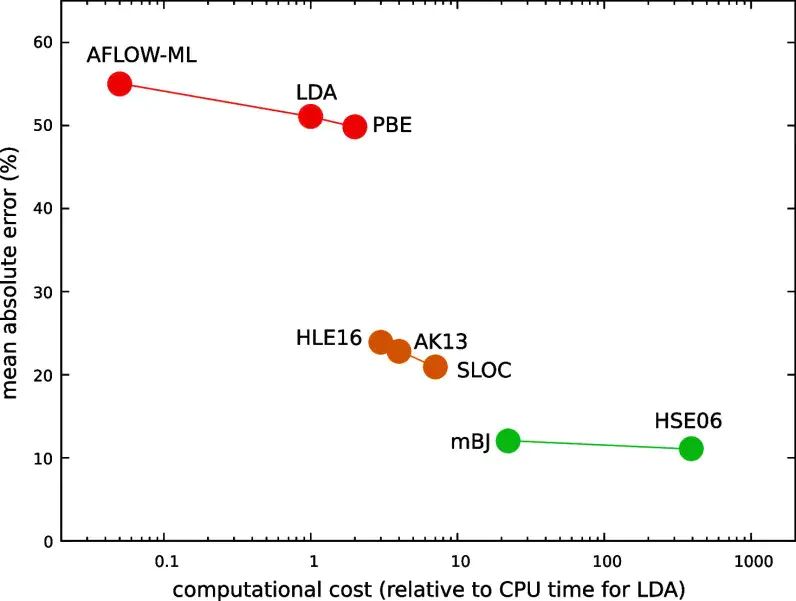
图5:机器学习重参数化与带隙基准测试结果用于说明半局域泛函修正的适用范围。DOI:10.1038/s41524-026-02009-w。