:是领域核心方法,通过维持恒定,更真实模拟电化学反应,优于恒电荷法。
电化学势DFTDFTVASPQuantum ESPRESSO恒电势计算()是计算电化学领域的核心方法,其反映电化学界面行为。

从理论基础来看,恒电势计算根植于电化学势的定义:电化学势(μ̃)是化学势(μ)与静电势(φ)的结合,表达式为μ̃=μ+zFφ,其中z为电荷数,F为法拉第常数。
恒电势计算的一约束确保计算体系始终处于目标电极电位下,贴近实验中通过电化学工作站控制电势的真实场景。
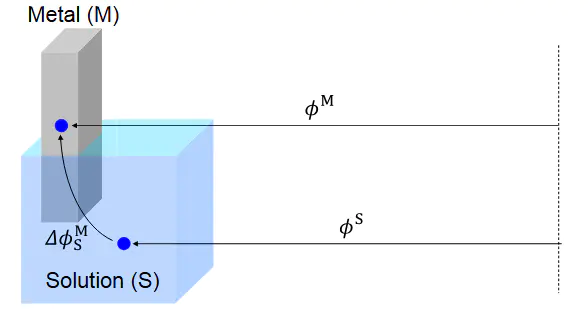
与传统的恒电荷计算相比,恒电势计算在模拟真实性和收敛性Q)进行计算,此时电极电势(U)会随体系状而恒电势法则直接控制电极电势(U),通过动态调整表面电荷分布抵消体系状态变化对电势的影响,更贴近实验中循环伏安法、计时电流法等控电势技术的实际条件。
从特性对比来看,恒电荷法控制变量为表面电荷(恒电势法控制变量为电极电势(),虽需迭代调整电荷分布导致计算复杂度升高,但收敛性更优,且能直接关联实验可观测量,因此成为现代电化学理论计算的主流方法。

恒电势计算的实现依赖于多套理论框架与算法创新,通过整合量子力学方法与电化学约束,实现对电极电势的精准控制。
第一类是()结合外场调控具体步骤为:首先构建包含电极表面、溶剂分子、电解质离子的电极溶液界面超胞模型,真空层厚度通常大于15 Å以避免周期性镜像电荷干扰;随后通过求解泊松方程计算体系静电势分布,明确电极相与溶液相的电势差;最后通过迭代调整表面电荷密度(σ),直至φ_solution-φ_metal等于目标电极电势(U)并收敛,这一过程需结合自洽场迭代确保电子密度与电势分布的一致性。
GC-DFT离子化学势与电极电势的条件下,通过巨配分函数描述体系的热力学平衡。
第三类是极溶液界面的双电层简化为电容(C),通过关系式ΔQ=C・ΔU快速估算反应能垒随电势的变化,虽精度较低,但计算效率极高,常用于初步筛选电势对反应路径的影响趋势。

关键算法突破为恒电势计算的高效实现提供了支撑。是核心,如等人提出的方法通过比较真空能级与溶液参考电位(如标准氢电极SHE)确定电势偏移量,动态调整表面电荷:每次迭代中,根据当前电势与目标值的偏差,通过线性响应理论估算所需的电荷调整量,逐步缩小误差直至收敛,该算法将电势收敛精度提升至±0.01 V以内。
通过引入位置依赖的介电常数(ε(r))近似溶液的极化效应,如VASPsol模块将电极表面附近的介电常数设为较低值,远离电极区域设为较高值,在降低显式溶剂分子计算成本的同时,较好地重现了双电层的静电势分布。
DOI:10.1103/PhysRevB.73.165402
具体计算工具
VASP用最广泛的第一性原理软件,通过参数控制体系偶极矩以消除层间静电相互作用,结合VASPsol模块实现隐式溶剂环境下的电势调控。
在中,VASP通过动态调整背景电荷密度平衡表面电荷,配合自洽场迭代确保电势收敛,其结果与实验的循环伏安曲线吻合度较高,是验证反应机理的可靠工具。

Quantum ESPRESSO作为开源模块化GC-DFT其模块化设计使得用户可根据需求组合不同的电子结构模块(如PWscf用于平面波计算、CP用于分子动力学),实现从静态电势控制到动态界面演化的多维度研究。
DOI:10.1088/0953-8984/21/39/395502
(Joint Density Functional Theory)Poisson-Boltzmann与其他软件相比,JDFTx在描述电极–溶液界面的静电相互作用时精度更高,其采用的自适应网格技术在电极表面附近加密网格,兼顾计算效率与界面区域的描述精度。
分子动力学DOI:10.1021/acs.chemrev.1c00675
以等人的论文《Dynamic potential–pH diagrams application to electrocatalysts for water oxidation》为例,该-pH为电催化氧析出反应()的催化剂活性预测提供了跨pH范围的理论框架。
-pH其步骤清晰且严谨:首先在DFT框架下固定电极电势(U),构建包含催化剂表面、关键中间体OER;最后通过方程将能垒转化为电流密度(j),即j=j₀exp (αnFη/RT),其中j₀为交换电流密度,α为传递系数,n为电子转移数,η为过电位(η=U-U°,U°为平衡电势),实现电流密度与电势、pH的定量关联。
IrO₂催化活性这一发现的物理机制在于:酸性条件下,IrO₂表面的中间体吸附能更接近理想值OER的能垒降低,从而降低过电位;而碱性条件下,表面OH⁻浓度升高导致OOH中间体吸附过强该研究的理论价值在于,DPPD为基催化剂的改性提供了明确方向——验证了恒电势计算在催化剂设计中的指导作用。

恒电势计算作为连接量子力学与宏观电化学现象的桥梁,其发展仍面临多尺度耦合、动态界面建模与效率提升等挑战,同时也在机器学习等新兴技术的推动下展现出广阔前景。
DFT计算虽能精准描述电子结构与化学键作用,但难以涵盖介观尺度两者的整合需要开发统一的多尺度理论框架——例如,通过将DFT计算的表面电荷密度作为边界条件输入介观模型,或采用粗粒化方法将原子级信息映射为介观参数,实现从电子转移到宏观电流的跨尺度关联,但目前此类方法的精度与效率仍需提升。
电势计算多采用静态模型,难以捕捉溶剂分子的动力学行为、离子的迁移扩散及电极表面的动态重构对电势分布的影响。
AIMD机器学习的引入为恒电势计算的效率提升提供了新路径。神经网络可通过训练表面电荷密度与中间体吸附能,将电势迭代过程的计算成本降低1-2个数量级。
总体而言,的成熟将实现“从电子结构到电池性能” 的直接预测,为电化学能源转化、电合成等领域的技术突破提供坚实的理论基础。