X射线吸收光谱XAS关于特定原子周围局部原子环境的精确信息测定原子间距离与依赖于长程有序结构的晶体衍射技术不同,XAS对样品的晶态、非晶态、液态或气态没有限制,使其在催化、材料科学、生物化学等领域得到广泛应用。
华算科技EXAFS基本物理原理实现对吸收原子与近邻原子间距离的精确测定XAS测量原子间距的核心原理根植于量子力学中的波粒二象性。
这一过程导致(Absorption Edge)。
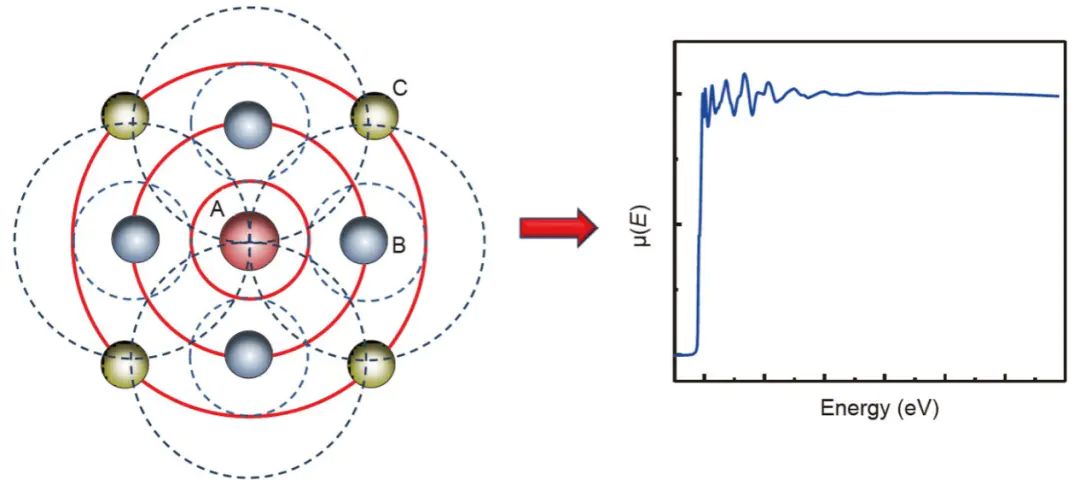
这些振荡并非随机的噪声,而是包含了丰富结构信息的信号。1.光电子波的产生与传播:2.邻近原子的散射:3.波的干涉:4.吸收概率的调制:由于入射X射线的能量在连续变化,光电子的能量和波长(λ)也随之改变。因此,简而言之,EXAFS谱图中的振荡,本质上是光电子波自身干涉效应的宏观体现,而振荡的频率直接与中心原子到邻近原子的距离相关。
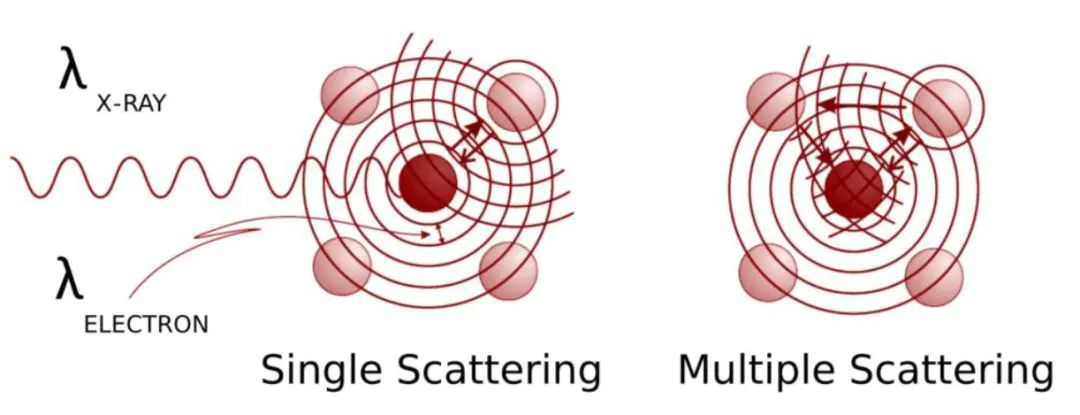
02、数据分析流程:从实验谱图到原子间距
首先,原始的吸收谱数据μ(E)(吸收系数随能量E的变化)需要进行标准化处理,包括预边扣除和归一化。接着,该振荡信号χ(E)通常会从能量空间(E)转换到光电子波矢空间(k空间),表示为χ(k),其中k是光电子的波数,与光电子的动能直接相关。
傅里叶变换:从k空间到R空间
傅里叶变换可以将信号从频率域(k空间)转换到距离域(R空间),得到一个径向分布函数(Radial Distribution Function, RDF)。
例如,第一个主峰通常对应于离中心原子最近的第一配位层,第二个峰对应第二配位层,以此类推。
距离的精确确定:相位校正与曲线拟合
这个相移包括了中心吸收原子和散射原子对光电子波产生的相位改变。因此,傅里叶变换得到的峰位会比真实距离小一个固定的偏移量,通常在-0.4至-0.5Å之间。为了获得精确的原子间距,必须进行相位校正。现代EXAFS分析通常采用曲线拟合的方法。
然后,通过最小二乘法等算法,将理论信号与实验信号进行拟合,不断调整模型中的结构参数(R,N,无序度因子等),直到理论曲线与实验数据达到最佳匹配。最终拟合得到的R值,即为高度精确的原子间距。
03、精度、分辨率与主要影响因素
典型精度与分辨率
0.02 Å在理想情况下,精度甚至可以优于。距离参数通常是EXAFS分析中最可靠的结构参数。
ΔR=π/(2kₘₐₓ)k值范围越宽(即EXAFS数据采集的能量范围越广),分辨率就越高。但在实际测量中,由于多种因素限制,kₘₐₓ往往受限,导致实际的分辨率通常被限制在之间。
主要影响因素
信号的信噪比(SNR)是决定测量精度的关键。高质量的EXAFS分析通常要求SNR优于1000。低信噪比会掩盖微弱的振荡特征,增加拟合的不确定性,从而降低距离测定的精度。
如分辨率公式所示,kₘₐₓ直接决定了分辨率。在实际实验中,kₘₐₓ可能受到样品中存在的其他元素的吸收边的限制。例如,当研究一个元素时,如果其能量更高的位置紧邻着另一个元素的吸收边,那么数据采集就必须在此中断,从而限制了kₘₐₓ。
EXAFS难以区分原子序数相近的散射原子,例如碳(C)、氮(N)和氧(O),或者铁(Fe)和锰(Mn)。这是因为它们的散射行为非常相似。
对于具有多个配位层或者配位环境高度无序的复杂体系,不同配位层的信号会相互叠加,使得数据分析和拟合变得更加困难,从而降低测定精度。
04、结论
从量子力学的干涉原理,到严谨的傅里叶变换和,EXAFS将微观世界的结构信息转化为了可精确解读的数据。