在上一篇推文《单细胞测序分析(四)| 数据标准化》中,我们探讨了如何通过标准化消除技术噪音,让不同细胞的基因表达量具有可比性。本期我们将继续深入,解锁单细胞分析的核心步骤——降维聚类与数据整合,带您从高维数据中挖掘生物学意义!
为什么需要降维?单细胞转录组数据通常有数千个基因的表达值,这使得数据非常高维。降维分析的目的是将这些高维数据映射到一个低维空间中,以便:
去除噪声:减少不必要的技术噪声,并保留重要的生物学信息。
可视化:将数据投射到二维或三维空间中,便于观察不同细胞群之间的差异和模式。
聚类和分类:在低维空间中,可以更好地进行细胞群的聚类或分类。
加速计算:降维后,后续的计算任务(如聚类或拟合模型)可以在低维数据上完成,减少计算量。
三种常用降维方法方法类型适用场景优点缺点PCA线性降维初步降维,数据预处理计算快速,易解释,适合大规模数据处理只能捕捉线性关系,难以处理复杂非线性数据t-SNE非线性降维数据可视化,局部结构分析展示复杂的非线性结构,常用于细胞群的可视化计算较慢,不适合大数据集,结果不稳定(非确定性)UMAP非线性降维数据可视化,局部和全局结构计算速度快,比 t-SNE 更稳定,能够保持全局和局部结构,适合大规模数据集对参数较为敏感,结果受超参数调整影响,需要调参优化实操# PCA 是基于 HVGseob <- RunPCA(seob, assay ="SCT")# s-SNE 不是基于表达矩阵做的,而是基于PCA结果seob <- RunTSNE(seob, assay ="SCT", dims = 1:30)# UMAPseob <- RunUMAP(seob, assay ="SCT", dims = 1:30)#可视化p_pca <- DimPlot(seob,reduction ="pca",# pca, umap, tsnegroup.by ="samples",label = F)p_umap <- DimPlot(seob,reduction ="umap",# pca, umap, tsnegroup.by ="samples",label = F)p_pca + p_umap
数据整合去批次效应方法原理如果存在明显的批次效应,则需要去除。
参考:https://satijalab.org/seurat/articles/seurat5_integration
五种方法: CCAIntegration, RPCAIntegration, HarmonyIntegration, FastMNNIntegration, scVIIntegration
数据规模:对于较大数据集,RPCAIntegration、HarmonyIntegration 和 FastMNNIntegration 更为合适。对于超大规模数据,scVIIntegration 表现优异。
模态类型:CCAIntegration 和 scVIIntegration 更适合处理多模态数据,如 RNA 和 ATAC-seq。
批次效应:HarmonyIntegration 和 FastMNNIntegration 是处理批次效应的好选择。
计算资源:如果计算资源有限,RPCAIntegration 是一个高效的选择,而 scVIIntegration 适合在有 GPU 资源时使用。
FastMNNIntegration 与 SCTransform 不兼容,需要用传统标准化方法 scVIIntegration 需要GPU 加速。
实操seob_harmony <- IntegrateLayers(object = seob,normalization.method ="SCT",orig.reduction ="pca",method = HarmonyIntegration,new.reduction ="integrated.harmony")#重新降维seob_harmony <- RunUMAP(seob_harmony, assay ="SCT", dims = 1:30,reduction ="integrated.harmony")DimPlot(seob_harmony,reduction ="umap",group.by ="samples",label = F)

tips:是否要进行批次整合,根据项目的具体情况。有时候整合后会丢掉真实的样本差异。
聚类分析FindNeighbors 函数用于计算数据集中每个细胞的 k 个最近邻居,并可选构建共享近邻图(SNN),通过计算每个细胞和其 k 个最近邻居之间的邻域重叠(Jaccard 指数)来实现。近邻图和共享近邻图在后续的聚类分析中用于确定细胞之间的相似性。
seob <- FindNeighbors(seob, reduction ="pca", dims = 1:20)
k.param:用于最近邻居算法的 k 值,默认为 20,表示计算每个细胞的 20 个最近邻居。
nn.method:用于寻找最近邻居的算法,选项包括 rann 和 annoy。annoy 是一种近似最近邻居算法,适合大规模数据集。
dims:用于构建近邻图的降维结果的维度,通常是 PCA 结果的前几个主成分。
FindClusters 函数用于基于共享近邻图的模块性优化算法(如 Louvain 或 Leiden 算法)来识别细胞簇。它首先计算每个细胞的 k 近邻,并基于近邻图构建 SNN 图,随后通过优化模块化函数来划分细胞簇。
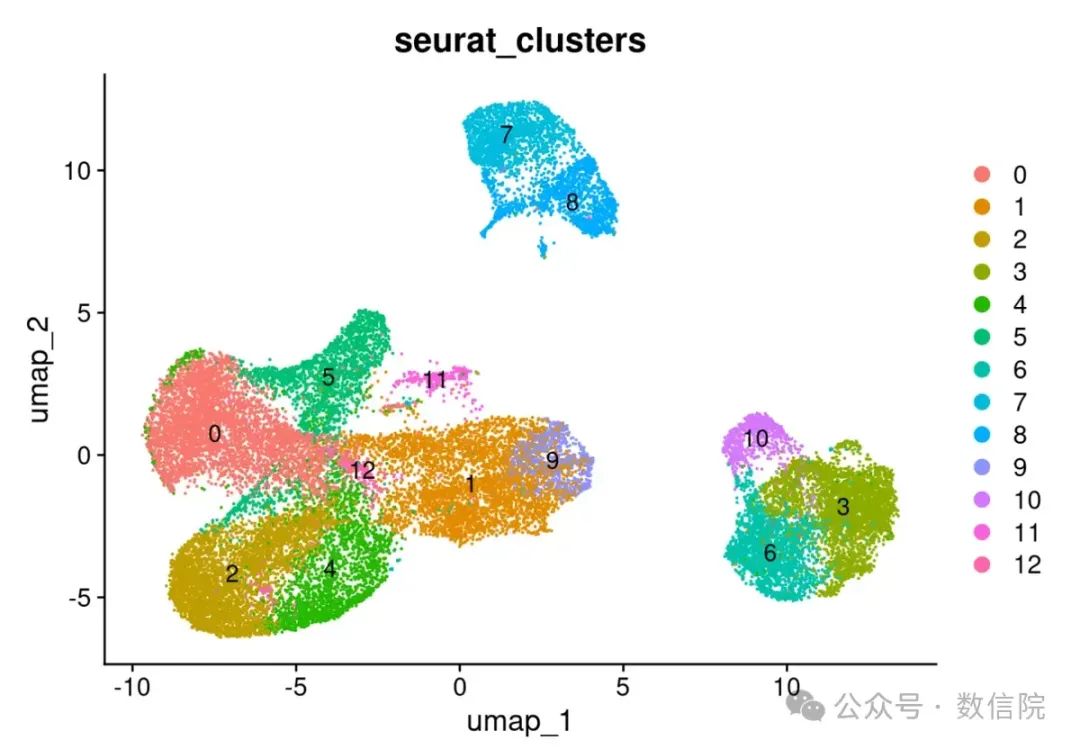
seob <- FindClusters(seob,resolution = 0.4,# 值越大,cluster 越多random.seed = 1)DimPlot(seob,reduction ="umap",group.by = c("seurat_clusters"),label = T)
resolution:控制聚类的分辨率。分辨率值越高,得到的细胞簇越多。
algorithm:用于模块化优化的算法,选项包括 Louvain(默认)和 Leiden(更适合大数据集)。Leiden 需要安装 leidenalg Python 包。