说明:本文主要介绍掺杂在材料计算中的基本含义、模型来源、电子结构响应和综合判断方式。
一、掺杂的基本定义:把外来原子放进晶体的什么位置?
掺杂指外来元素或非本征缺陷进入材料晶格后,改变局部配位、价电子分布和能级结构的过程。它并不等同于在化学式里多写一个元素名,核心在于 掺杂原子占据了哪类晶格位置、周围键长怎样重排、体系为维持电荷平衡产生怎样的电子或空穴响应。对于半导体和催化材料,掺杂常被用来调节载流子浓度、缺陷能级、表面吸附强度、界面电场和光吸收范围;这些变化有时来自局部化学键,有时来自长程静电势的移动。
最常见的几类位置包括 替位掺杂、间隙掺杂和缺陷复合掺杂。替位掺杂是外来原子替换晶格中的某个本征原子,例如用碱土金属替换 MoS2 中的 Mo 位;间隙掺杂是小原子进入晶格空隙;缺陷复合掺杂则把外来原子和空位、反位缺陷或电荷补偿缺陷连在一起讨论。若掺杂浓度很高,体系可能更接近合金、固溶体或有序化合物;若浓度很低,单个掺杂中心的局部结构和统计分布会变得更敏感。DFT 超胞模型里的“一个掺杂原子”本质上对应周期重复的掺杂阵列,超胞大小直接决定名义掺杂浓度,也决定掺杂中心之间是否存在人为相互作用。
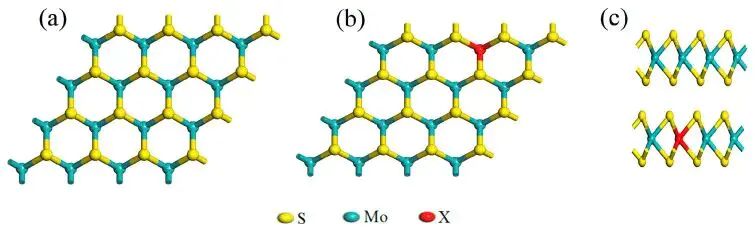
图1. MoS2 单层及 Be、Mg、Ca、Sr、Ba 替位掺杂模型的俯视和侧视结构,红色 X 表示掺杂原子。DOI:10.3390/molecules28166122
把 主体材料、掺杂原子、缺陷位点和电荷态 分开后,掺杂对象的差别会直接转化为不同物理含义。主体材料是晶体骨架,掺杂原子是进入骨架的外来成分,缺陷位点是它所占据的具体位置,电荷态描述缺陷与电子库之间的电子交换。把这几个概念分开后,才能理解为什么同一种元素掺入不同位点会产生相反效果:同样是 N 掺杂,进入氧化物氧位、进入碳材料石墨位、吸附在表面空位附近,对能带、磁矩和表面反应中间体的影响完全不同。元素身份只是第一层信息,位点和配位环境才是掺杂效应的结构来源。
二、第一性原理如何描述掺杂是否可能存在?
在第一性原理语境里,掺杂是否能够稳定进入晶格,首先要回到 原子库和晶体骨架之间的能量竞争。体系能不能容纳某个外来原子,通常用缺陷形成能描述。形成能把掺杂后超胞总能、未掺杂超胞总能、被移走或加入原子的化学势、费米能级位置和电荷修正项联系起来。中性替位掺杂的表达较直观:用含掺杂原子的超胞能量减去本征超胞能量,再减去加入掺杂原子的化学势,加上被替换原子的化学势。带电缺陷则需加入 q(EF + EVBM) 一类项,反映缺陷和电子库之间的电子交换。
形成能会随着 元素化学势窗口 改变,同一个掺杂位点在富金属、富非金属、富氧或贫氧条件下可能呈现不同热力学偏好,进而改变哪种掺杂位点更容易出现。对于替位掺杂,掺杂原子半径、价态和电负性决定局部应变与电荷补偿代价;对于间隙掺杂,晶格空隙大小和局部静电势更敏感。计算中常比较多个位点的形成能,寻找低能掺杂构型,但 低形成能只说明热力学上更容易形成,并不自动代表材料性能更好。
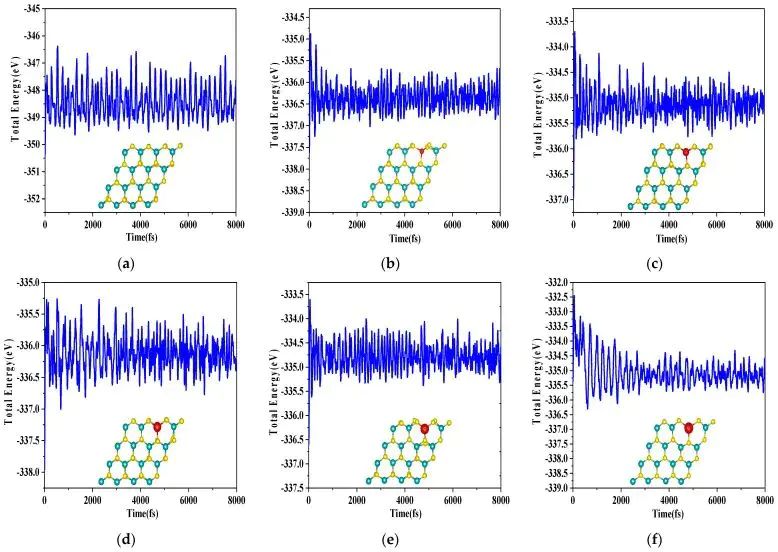
图2. MoS2 及碱土金属替位掺杂 MoS2 在 300 K AIMD 中的能量波动和末态结构。DOI:10.3390/molecules28166122
结构优化和有限温度动力学补充的是另一层信息。优化结构告诉我们掺杂中心附近的键长、键角和层间起伏如何松弛;AIMD 则观察一定温度下是否出现明显断键、扩散或重构。若形成能较低但局部结构在热扰动下迅速塌陷,掺杂位点难以作为稳定功能中心;若结构稳定但形成能很高,合成时可能需要非平衡条件、等离子体处理或后退火才能引入。形成能、优化构型和 AIMD 共同限定了掺杂“存在”的范围。
掺杂计算还需区分化学掺杂和载流子掺杂。化学掺杂把外来原子显式放入超胞,它会改变局部几何和轨道杂化;载流子掺杂常通过加电子、去电子或改变背景电荷模拟,它强调费米能级移动和电子填充变化。两者都能改变电导、磁性或反应能垒,但物理来源并不相同。把真实原子位点和纯电子填充分开,可以避免把结构畸变、电荷转移和费米能级移动混成一个概念。
三、掺杂为什么会改变能带、态密度和局部电荷?
掺杂改变电子结构,首先来自 局部势场和价电子数 的变化。外来原子的核电荷、电负性和价轨道能级不同于主体原子,进入晶格后会重新分配邻近原子的电子密度,局部势垒、键级和轨道重叠随之改变。若掺杂原子的价电子数高于被替换原子,体系可能出现额外电子并推动 EF 上移;若价电子数较低,还可能产生空穴或受主能级。施主掺杂和受主掺杂 的差别,本质上来自掺杂中心与主体能带之间的电子交换。
态密度给出的是能量空间里的电子态分布。总 DOS 显示材料在各能量位置有多少可占据或未占据态,PDOS 进一步把这些态投影到元素和轨道上。掺杂后若带隙内出现尖锐峰,常提示局域缺陷态;若导带底或价带顶附近的新态由掺杂原子和邻近原子共同贡献,说明发生了轨道杂化。对于催化材料,表面金属 d 态、反应中间体 p 态或吸附原子 s 态的重叠会影响吸附键强度;对于半导体,带边组成和缺陷能级深浅 决定载流子是容易被激发,还是容易被俘获。
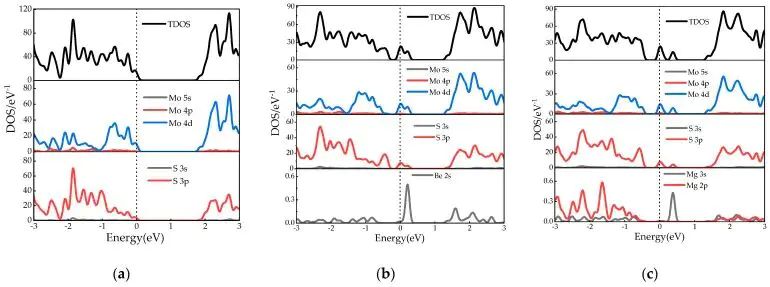
图3. 本征 MoS2、Be-MoS2 和 Mg-MoS2 的总态密度与分波态密度。DOI:10.3390/molecules28166122
如果把 Be、Mg 与 Ca、Sr、Ba 分开比较,DOS/PDOS 的重点不只是曲线形状是否相似,还在于掺杂原子半径增大后,局部畸变会怎样改变 Mo-d、S-p 与掺杂原子价轨道之间的混合。较大半径元素带来的晶格起伏通常更明显,带边附近态密度的分布位置和峰宽会随配位环境变化。同一族元素的连续比较 能把元素尺寸、轨道能级和缺陷态变化联系起来,图4对应这组更大半径掺杂元素。
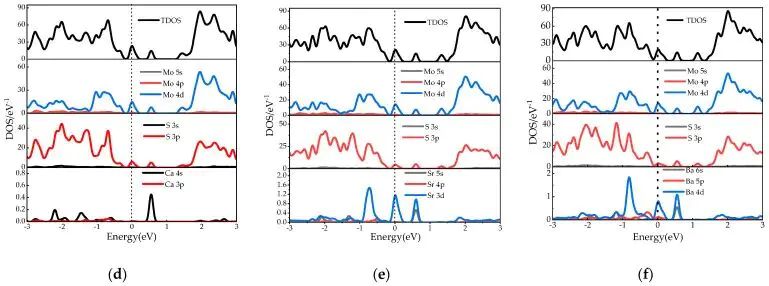
图4. Ca-MoS2、Sr-MoS2 和 Ba-MoS2 的总态密度与分波态密度。DOI:10.3390/molecules28166122
电荷分析把电子结构变化转回实空间。差分电荷密度能标出电子在掺杂中心周围富集或耗尽的位置,Bader 电荷能给出不同原子盆之间的电子分配趋势,电子局域函数可以辅助判断共价性增强还是离子性增强。需要注意的是,电荷转移的方向和大小不能单独决定性能;它只能说明局部键合和静电环境怎样变化,仍需与能带、吸附构型、反应路径或载流子寿命共同判断。
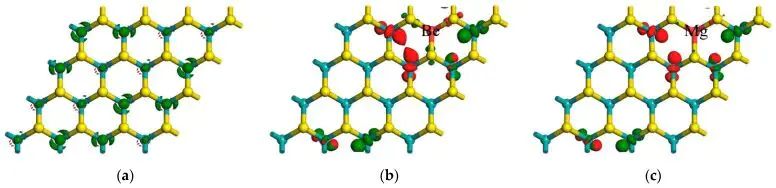
图5. 本征 MoS2、Be-MoS2 和 Mg-MoS2 的 LUMO 与 HOMO 空间分布。DOI:10.3390/molecules28166122
HOMO、LUMO 或带边电荷密度图强调的是波函数空间分布。掺杂若让 LUMO 更集中在某个金属位附近,电子注入后该位置更容易参与还原过程;若 HOMO 分布靠近非金属骨架,空穴氧化过程可能更受该区域控制。空间分布并不直接等同于反应速率,却能提示载流子、吸附物和活性位点是否处在同一局部环境。电子态的能量位置和空间位置 必须同时进入判断,掺杂效应才不会被简化成一个带隙数字。
四、怎样判断掺杂带来的性能变化?
掺杂对性能的影响往往牵涉 结构稳定、能级位置和界面电荷交换,不能用单向度变化概括。带隙变窄可能扩大可见光吸收范围,却还可能引入复合中心;费米能级移动可能提高电导,却可能改变界面能级匹配;表面吸附变强有利于某些中间体活化,却可能让产物脱附变慢。材料计算需要把 结构稳定、电子结构、界面能级和目标反应 放入同一个物理图像中。只有当目标性质被明确后,掺杂改变的方向才有意义。
功函数是判断界面电子交换能力的常用指标。它表示电子从材料费米能级移到真空能级所需能量,受 EF、表面偶极和局部电荷重排共同影响。掺杂可能通过施主或受主行为改变 EF,也可能通过表面偶极改变 Evac。两种变化在数值上都会进入功函数,但对应的物理含义并不相同。若讨论光催化或电催化界面,功函数需与带边位置、溶液电势和吸附态电荷转移 联合分析。
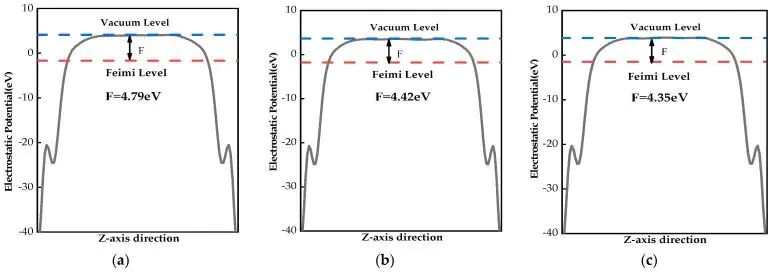
图6. 本征 MoS2、Be-MoS2 和 Mg-MoS2 的静电势分布与功函数。DOI:10.3390/molecules28166122
把功函数拆成两组图阅读时,前一组更适合观察本征、Be 和 Mg 掺杂之间的低半径替位差别;后一组则突出 Ca、Sr、Ba 半径增大后,表面静电势平台和真空能级相对位置的改变。功函数变化来自 EF 与 Evac 的相对移动,元素尺寸引起的表面偶极变化 会让同族掺杂呈现不同界面电子逸出代价,图7对应这一路径。
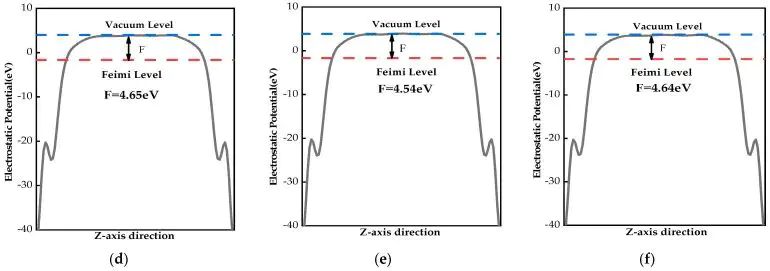
图7. Ca-MoS2、Sr-MoS2 和 Ba-MoS2 的静电势分布与功函数。DOI:10.3390/molecules28166122
若目标是光响应,吸收系数、介电函数、折射率和带边位置会比单个带隙值更有信息量。掺杂可以在可见光区域引入新的跃迁通道,也可以通过局部态造成非辐射复合。对于二维半导体,Sr 和 Ba 等较大原子掺杂时,局部结构畸变、静电势分布和费米能级位置可能同时改变,光吸收增强未必意味着光生载流子分离就更好。吸收强度、载流子迁移、复合倾向和反应位点 需要在同一材料目标下逐项核对。
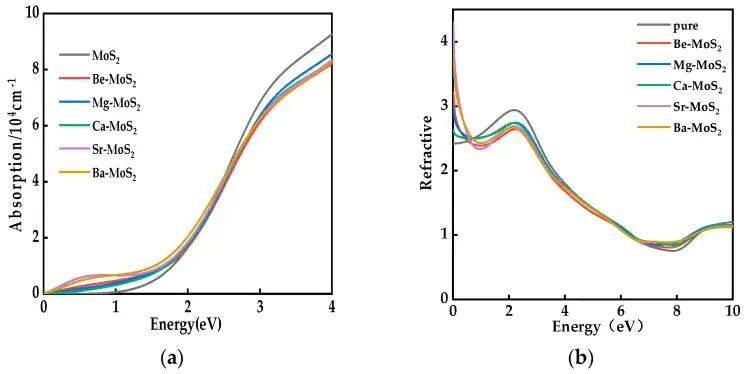
图8. 本征及碱土金属替位掺杂 MoS2 的光吸收曲线和折射率曲线。DOI:10.3390/molecules28166122
综合判断掺杂效应时,可以把问题拆成四个层次:外来原子能否稳定进入晶格,进入后是否形成明确局部配位,电子结构是否出现可解释的带边或缺陷态变化,目标性质是否沿着预期方向改善。若研究电催化,还需加入吸附自由能、过渡态能垒和溶剂/电势效应;若研究半导体器件,则关注载流子浓度、迁移率、深能级陷阱和接触势垒。掺杂是一种结构和电子态耦合的调控手段,它可能提高某个性能,也可能牺牲另一项性能。真正可靠的判断来自材料目标、掺杂位点和电子结构证据之间的相互一致。