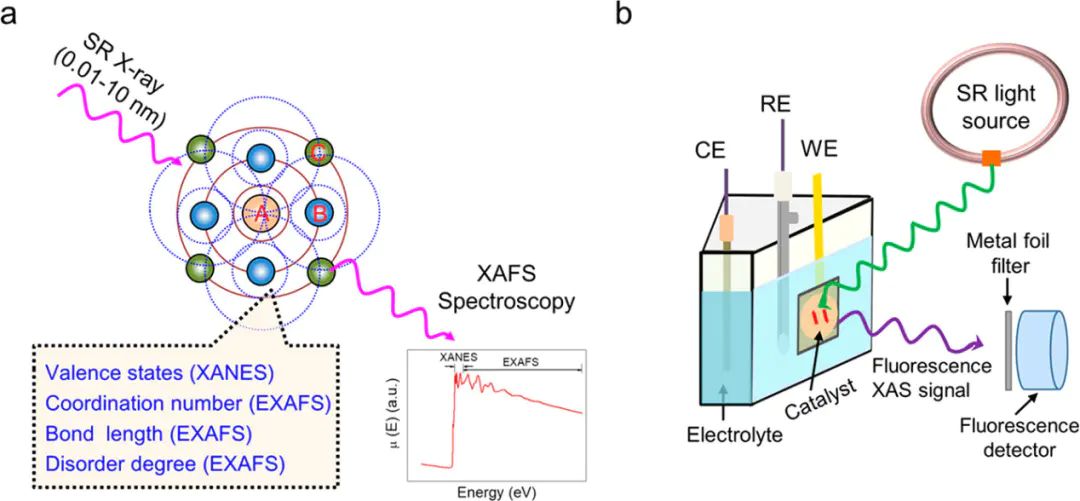
同步辐射义、“三高一广” 优势及技术原理,解决催化研究痛点的关键作用单原子催化剂XAFSFTIRDFT同步辐射光源单原子位点SAS得益于超高亮度和30–psXAFSKOHeV-4直接捕获孤立金属中心的氧化态漂移和配位数变化。
XCVXANESEXAFSXAFS这个波在与周围原子发生散射并返回中心原子的过程中,与入射X射线光子产生干涉,进而在X射线吸收系数中形成振荡结构,这就是XAFS现象的根源。
光谱分为X射线吸收近边结构(XANES)和扩展X射线吸收精细结构(EXAFS)两个主要区域。
提供关于中心原子周围键长、配位数和结构无序度的丰富结构信息。
为了收集高质量的XAFS数据,必须综合考虑X射线源、探测器和检测模式以及样品制备等多方面因素。
XAFS通过这种技术,可以实时观察,从而更好地理解催化剂的结构与性能之间的内在联系。这种深入理解,不仅能优化现有催化剂的性能,还能为设计和制备新型催化剂提供重要的理论依据。
同步辐射优势
高亮度:10-10倍,能探测到材料中极微量的成分(比如单原子催化剂中仅wt%高准直性:高偏振性:波长范围广:X正是这些优势,让同步辐射能够在原位条件(即催化剂工作的真实反应状态)下,实时追踪,而这恰恰是传统表征技术(如常规X射线衍射、电镜)难以实现的。
催化研究为什么需要依赖同步辐射?
动态变化:原子级分散:N电子态敏感:Co²+而同步辐射的出现,恰好解决了这些“痛点”。特别是X射线吸收光谱(XAS)技术(包括XANES和EXAFS两个部分),更是成为催化研究的不可或缺的一环——前者用于,后者用于。
实际应用案例
X构演变。(DOI:10.1038/s41929-018-0203-5)
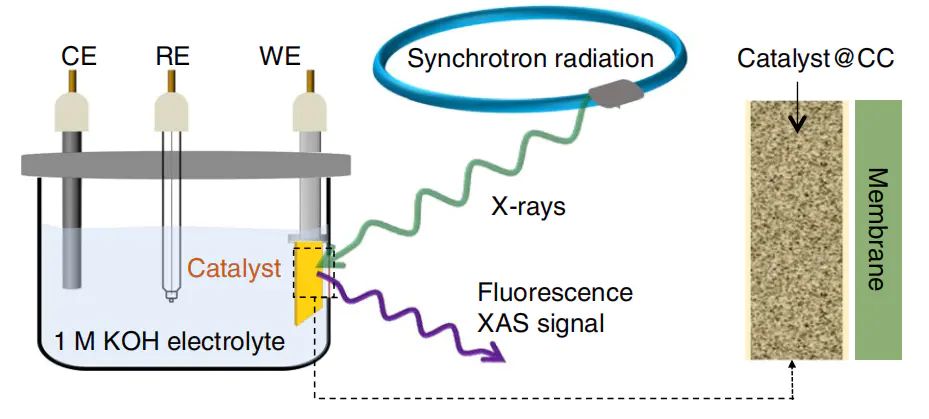
揭示Co/PCN催化剂在HER过程中的通过原位装置测试K-edgeXAS1在碱性HER过程中的电子与配位动态。
0.10VeV34CoOOH2.200.04V差谱(图b)进一步证实边前–边后特征强度变化源于Co空轨道增加而非背景漂移。
NO2在单原子Co位点吸附并诱导形成高价Co(2关键电子结构基础EXAFSN构型(,RCo–ÅNO1N–1CN=3N=2.01,RCo–Å当电位降至−0.04V时,配位层进一步调整为Co–2O(,RCo–ÅO=1.98),其中新增O原子源自HOCo–2HO(HOCo–2共吸附的高价态结构,该构型即碱性HER的活性中心。k³χ(k)振荡拟合(图b)与实验曲线高度重合(R因子),验证了上述模型的可靠性。
3500cm处仅于1出现显著增宽峰,归属为Co–伸缩振动,排除物理吸附HOCo2,3XAS30.2,定量证实Co空轨道增多、氧化态升高,与XANES结果自洽。综上,–2逐步配位诱导的Co–2O高价单元是驱动碱性的本征活性构型nm图d的元素分布图显示C、N、P与Co信号在相同区域均匀重叠,磷来源于PCN载体的磷化改性,且未出现Co-富集畴区,与STEM结果互为印证。
1仅需89 mV过电位即可输出10 mA cm几何电流密度,性能优于空白与Co/PCN-1VolmerHeyrovsky上述结果共同确立Co/PCN原位XAFS追踪结构演变提供可靠基准DFT理论研究HOCo/PCNEXAFSCo–26×6溶剂效应由显式HO+。
a2吸附→II.O–断裂(Volmer)→III.H2形成HO⁺-like**复合→V.H脱附,其中第二步被确认为速率决定步骤()。
Co表面对2的吸附能为0.94,比Pt(111)的0.46高0.48eV,表明更强的水捕获能力;然而该步骤放热0.31,熵校正后仍自发。
Co上仅为eVeV0.28,说明O–断裂既容易又放热,可持续、快速地向表面供应质子。
3d2p1.0区间出现明显轨道重叠,电子从Co向吸附OH转移,分析给出Co氧化态由初始+2.05升至+2.16,与XANES实验值(+2.20→+2.40)趋势一致。
Co–2Volmer实现碱性HER活性超越Pt的源头优势同步辐射技术(尤其)对原子级结构与电子态灵敏度高,在单原子催化剂活性位点动态研究中优势突出。
EXAFS不过,其在复杂体系中存在信号干扰、瞬态结构难捕捉等问题。未来,随着光源性能提升、多技术联用及解析方法优化,将更精准助力单原子催化剂设计,为能源转化、碳中和等领域提供支撑。