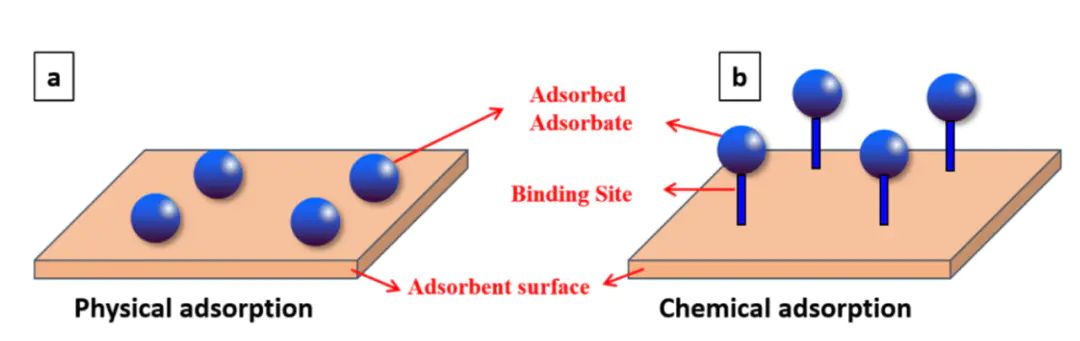
吸附。吸附可以发生在气体、液体与固体之间,常见于催化、传感、污染物去除与电极界面等场景。
主要由范德华力、静电力等弱相互作用驱动,能量较小,可逆;
则涉及化学键的形成,伴随电子重排与较高的结合能,通常更强也更具选择性。
。
10.3390/w14203203
包括分子运移到表面、初步捕获、重排或解离以及可能的反应与脱附,对来说,把吸附想象为分子与表面之间的一场“”:有时候只是握握手(物理吸附),有时则谈成合同要长期合作(化学吸附)。
从计算化学看吸附过程
为理解吸附机理提供了原子级的“放大镜”。
密度泛函理论,它可以计算吸附能、几何构型、电子态变化以及吸附后可能出现的活性中间体。
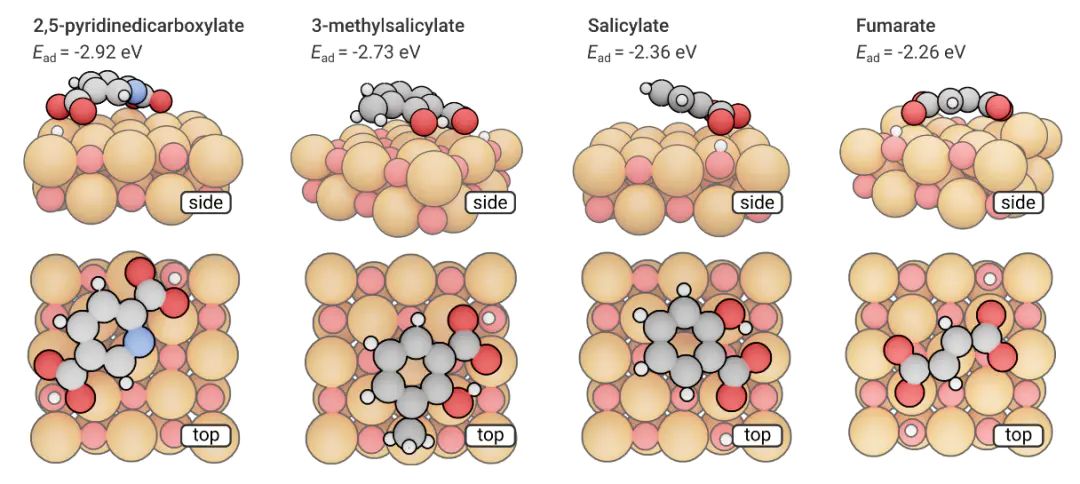
,研究者可以预测分子最可能“停靠”的位置;通过态密度和电荷分析下图展示了四种小有机分子在部分羟基化MgO(100)表面的DFT计算吸附能,包含分子结构侧视图、顶视图和具体吸附能数值。
10.1016/j.electacta.2020.136166
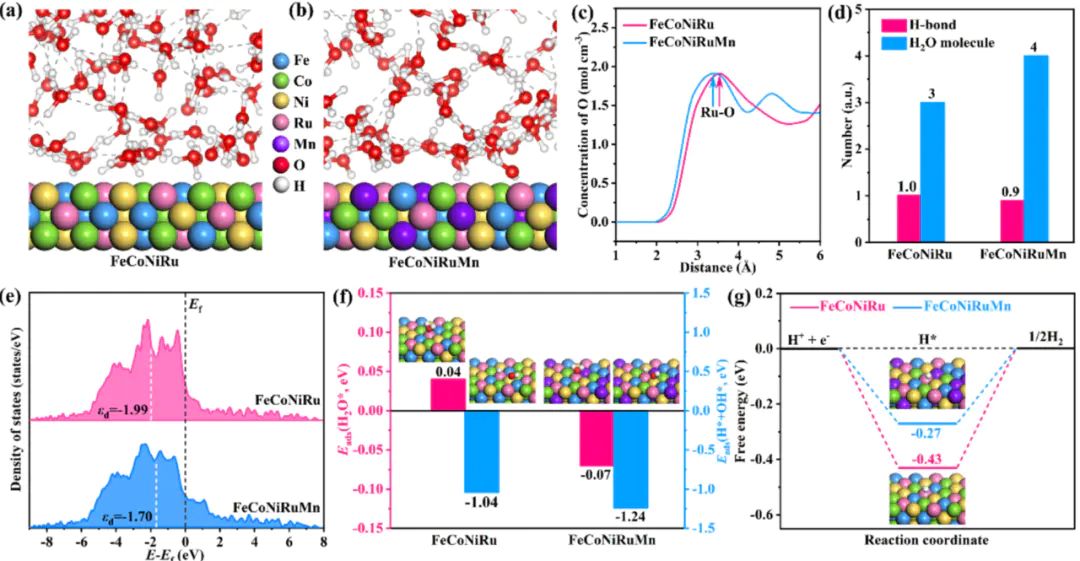
,从而估算速率常数。若要考虑温度、溶剂或界面重构的影响,分子动力学(MD或AIMD)AIMD模拟。界面水浓度更高且氢键网络更无序,促进了H2O*/OH-的传输。
表明,FeCoNiMnRu的d带中心更接近费米能级,优化了中间体吸附。。FeCoNiMnRu HEA具有更理想的氢吸附自由能,意味着更高的本征HER活性。
10.1016/j.cej.2025.161070
建议
吸附机理。计算能帮助筛选活性位点、设计表面修饰或辅助剂来提高选择性与活性。
建议1)首先明确研究目标(热力学优先还是动力学优先),选择合适的模型尺寸与表面代表性;
DFT3)对于含溶剂或电极电位的体系,考虑AIMD或QM/MM以及恒电势模拟以逼近实验条件;
通过严谨的建模与多方法验证,计算化学可以把“表面上发生什么”变成可量化的机理解释,直接推动材料与工艺的优化。
总结
是表面科学的核心,从分子到材料性能都受其支配。
热力学与动力学DFT实践中需注意模型代表性、泛函和参数敏感性以及溶剂与界面条件的处理合理的计算工作流程